Lys417 acts as a molecular switch that regulates the conformation of SARS-CoV-2 spike protein
Abstract
SARS-CoV-2 spike protein plays a key role in mediating viral entry and inducing host immune responses. It can adopt either an open or closed conformation based on the position of its receptor-binding domain (RBD). It is yet unclear what causes these conformational changes or how they influence the spike’s functions. Here, we show that Lys417 in the RBD plays dual roles in the spike’s structure: it stabilizes the closed conformation of the trimeric spike by mediating inter-spike–subunit interactions; it also directly interacts with ACE2 receptor. Hence, a K417V mutation has opposing effects on the spike’s function: it opens up the spike for better ACE2 binding while weakening the RBD’s direct binding to ACE2. The net outcomes of this mutation are to allow the spike to bind ACE2 with higher probability and mediate viral entry more efficiently, but become more exposed to neutralizing antibodies. Given that residue 417 has been a viral mutational hotspot, SARS-CoV-2 may have been evolving to strike a balance between infection potency and immune evasion, contributing to its pandemic spread.
Editor's evaluation
In this valuable study, Li et al. identify a molecular switch within the SARS-CoV-1/2 spike proteins, in position 417. Based on comparisons between CoV-1 and CoV-2 sequences, the authors propose that the nature of the residue of this position determines whether the spike favors a closed or an open conformation. The authors present solid cryo-EM and biochemical data in support of their hypothesis. The paper will be of general interest to those interested in molecular mechanisms of viral infection by SARS-CoV-2 and other coronaviruses.
https://doi.org/10.7554/eLife.74060.sa0Introduction
Coronaviruses have long infected humans, yet none caused the same devastation as SARS-CoV-2 has (Li et al., 2020; Huang et al., 2020). For instance, a virulent and lethal coronavirus, SARS-CoV-1, caused a much smaller outbreak in 2002–2003 (Lee et al., 2003; Peiris et al., 2003). Numerous human coronaviruses such as NL63-CoV cause common colds annually (Fouchier et al., 2004; van der Hoek et al., 2004). With an intermediate virulence, SARS-CoV-2 causes a fatality rate that is significantly lower than that of SARS-CoV-1, but much higher than that of NL63-CoV. Because of its intermediate virulence, SARS-CoV-2 carriers show clinical signs that facilitate the spread of the virus: they may develop mild or no symptoms, experience delayed onset of symptoms, develop low levels of neutralizing antibodies, or endure prolonged virus shedding period (Wu et al., 2020; Zhou et al., 2020; Wölfel et al., 2020; Gao et al., 2020; Kronbichler et al., 2020). These features contribute to the wide spread of SARS-CoV-2 and severe health outcomes, triggering a global COVID-19 pandemic that was unprecedented in the era of modern medicine. Understanding the molecular determinants of COVID-19 is crucial for comprehending the recent pandemic and preventing future ones.
SARS-CoV-2 spike protein played a central role in the COVID-19 pandemic. It guides viral entry into host cells and is also a major target for the host immune responses (Du et al., 2009; Li, 2016). On newly packaged virus particles, the trimeric spike protein has a pre-fusion structure in which three receptor-binding S1 subunits sit on top of a trimeric membrane-fusion S2 stalk (Figure 1A). During viral entry, a receptor-binding domain (RBD) in S1 binds to a receptor on host cell surface for viral attachment and S2 switches to a post-fusion structure for the fusion of viral and host membranes (Li, 2016; Li, 2015). SARS-CoV-2, SARS-CoV-1, and NL63-CoV can all use ACE2 as their receptor (Li et al., 2003; Li et al., 2005; Shang et al., 2020c; Wan et al., 2020a; Wu et al., 2009). Early in the pandemic, we described three distinct characteristics of SARS-CoV-2 spike: its exceptional binding affinity for human ACE2, its proteolytic activation by human protease furin, and its existence as a mixture of two conformations: an open conformation in which the RBD is exposed and accessible to ACE2 and a closed conformation in which the RBD is buried and inaccessible to ACE2 (Shang et al., 2020b). While much has been learned about the first two characteristics (Shang et al., 2020c; Lan et al., 2020; Essalmani et al., 2022; Peacock et al., 2021; Geng et al., 2022; Zhang et al., 2023), the third still remains poorly understood. Interestingly, SARS-CoV-1 spike is predominantly in the open conformation, whereas NL63-CoV spike remains closed (Walls et al., 2016; Wrobel et al., 2020; Gui et al., 2017). Thus, two key questions arise: what molecular switches regulate the conformation of coronavirus spikes and how do the conformational changes influence coronaviruses’ infectivity and host immune responses?
Figure 1

Identification of residue 417 as a molecular switch that regulates the conformation of SARS-CoV-2 spike.
(A) Structure of trimeric SARS-CoV-2 spike ectodomain in the closed conformation with three receptor-binding domains (RBDs) down (PDB 6VXX). Each monomeric subunit of the spike trimer is colored differently. The RBD contains a core structure (in cyan) and a receptor-binding motif (RBM; in magenta). Lys417 in the RBD is shown as blue sticks. (B) A hydrogen bond is formed between the side chain of Lys417 from one spike subunit and the main chain of Asn370 from another spike subunit, stabilizing the trimeric spike in the closed conformation. (C) Residue 417 is a valine in SARS-CoV-1 spike and has been a mutational hotspot in later SARS-CoV-2 variants.
In this study, we used cryo-EM and biochemical approaches to identify spike residue 417 as a molecular switch that regulates the conformation of SARS-CoV-2 spike. We delved deeper into how this molecular switch affects receptor binding, viral entry, and immune evasion by SARS-CoV-2. Through regulation of its spike’s conformations, SARS-CoV-2 may have struck a balance between infection potency and immune evasiveness.
Results
To understand the molecular mechanism that controls the spike’s switching between open and closed conformations, we compared the sequences of SARS-CoV-2 and SARS-CoV-1 spikes in the context of their tertiary structures. We identified residue 417 as potentially a key difference between the two spikes: in the closed SARS-CoV-2 trimeric spike, Lys417 in the RBD from one spike subunit forms a hydrogen bond with the main chain of Asn370 in the RBD from another spike subunit, stabilizing the RBDs in the closed conformation (Walls et al., 2020; Figure 1B). In SARS-CoV-1 spike, however, this residue becomes a valine (Li et al., 2005; Shang et al., 2020a; Figure 1C), losing its capability to form the hydrogen bond with another spike subunit and potentially destabilizing the closed conformation of the spike. Thus, we hypothesized that a K417V mutation causes more SARS-CoV-2 spike molecules to take the open conformation. To test this hypothesis, we investigated the impact of the K417V mutation on the structure and function of SARS-CoV-2 spike using a combination of cryo-EM and biochemical approaches.
To examine how the K417V mutation affects the conformation of SARS-CoV-2 spike, we performed cryo-EM analysis of the RBD conformation of SARS-CoV-2 spike ectodomain containing either Lys417 or Val417. To this end, we introduced the K417V mutation into SARS-CoV-2 spike ectodomain and purified both versions of the protein: K417-spike containing Lys417 and V417-spike containing Val417. Due to the lack of the transmembrane anchor, the recombinant spike ectodomain was intrinsically unstable. To stabilize the recombinant spike ectodomain, we introduced six proline mutations into its S2 subunit to lock up its pre-fusion structure, introduced mutations to its furin motif to prevent it from being cleaved during molecular maturation, and added a C-terminal foldon tag to facilitate its trimerization (Shang et al., 2020b; Hsieh et al., 2020; Ye et al., 2022). We successfully obtained the stabilized K417-spike and V417-spike and collected cryo-EM data on both proteins (Figure 2—figure supplements 1 and 2; Supplementary file 1). For the K417-spike, 55% of the molecules were in the closed conformation with all three RBDs down, whereas the rest of the molecules (45%) were in the open conformation with one RBD up and two RBDs down (Figure 2A and B). In contrast, for the V417-spike, these numbers became 32 and 68% for the closed and open conformations, respectively (Figure 2C and D). Therefore, the K417V mutation allowed more SARS-CoV-2 spike molecules to take the open conformation, confirming our hypothesis.
Figure 2 with 2 supplements see all
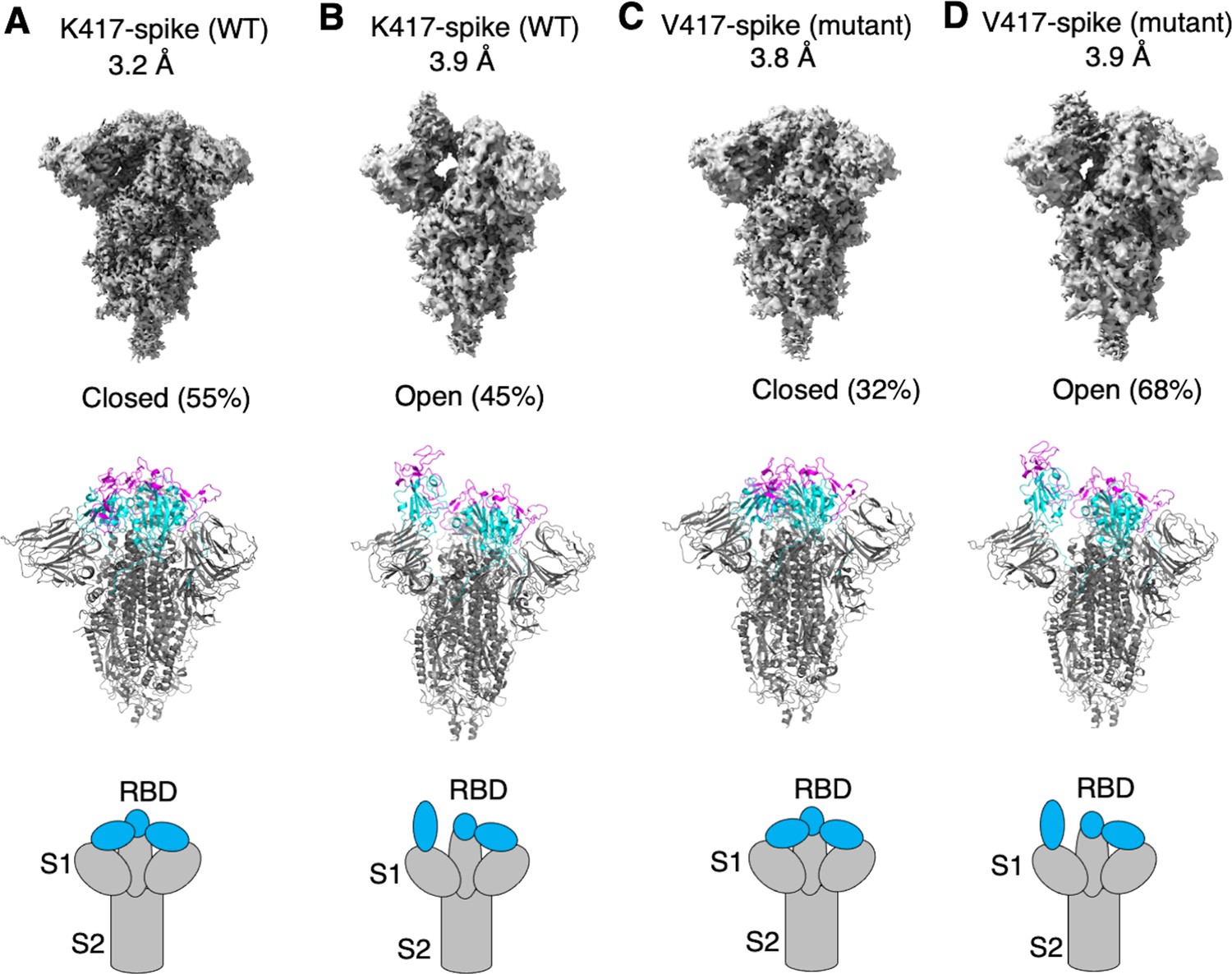
Cryo-EM analyses of residue 417 in regulating the conformation of recombinant SARS-CoV-2 spike ectodomain.
Prototypic SARS-CoV-2 spike ectodomains containing either Lys417 (as in prototypic SARS-CoV-2) or Val417 (as in SARS-CoV-1) were subjected to cryo-EM analyses and the particle distributions for open and closed conformations were calculated. Cryo-EM densities (top), atomic models (middle), and schematic presentations (bottom) for each of the protein classes are shown. (A) K417-spike in the closed conformation. (B) K417-spike in the open conformation. (C) V417-spike in the closed conformation. (D) V417-spike in the open conformation.
While the above cryo-EM approach examined the conformation of recombinant spike ectodomain, we further examined the impact of the K417V mutation on the membrane-anchored and full-length SARS-CoV-2 spike that do not contain S2 or furin motif mutations. It has been shown that ACE2 only binds to the standing-up RBD in the open spike (Gui et al., 2017; Ye et al., 2021), suggesting that the open spike conformation is necessary for ACE2 binding. Hence, we characterized the capabilities of K417-spike and V417-spike in binding ACE2. First, we expressed the K417-spike and V417-spike on human cell surface and performed a protein pull-down assay using recombinant human ACE2 as the bait and the cell-surface-anchored spikes as the target (Figure 3A). For cross-validation, both His-tagged ACE2 and Fc-tagged ACE2 were used. We previously showed that this pull-down assay is a reliable method to probe the RBD conformation of cell-surface-anchored spikes, with higher pull-down levels of spikes associated with more spike molecules in the open conformation (Shang et al., 2020b). Western blot analysis showed that some of the spike molecules had been cleaved at the S1/S2 boundary, confirming the intact furin motif (Figure 3A). The pull-down result showed that, compared to K417-spike, more V417-spike molecules had been pulled down by ACE2, suggesting that V417-spike had an increased probability of binding to ACE2 (Figure 3A). Second, we conducted a flow cytometry assay to detect the interactions between recombinant human ACE2 and cell-surface-anchored spikes (Figure 3B, Figure 3—figure supplement 1). Here, two versions of both K417-spike and V417-spike were used: one containing the intact furin motif and the other the inactivated furin motif. The result showed that V417-spike binds to ACE2 better than does K417-spike, no matter whether the furin motif was intact or inactivated (Figure 3B, Figure 3—figure supplement 1). Taken together, both protein pull-down and flow cytometry results reveal that the K417V mutation allowed more SARS-CoV-2 spike molecules to take the open conformation, confirming the cryo-EM data and our hypothesis.
Figure 3 with 1 supplement see all

Biochemical analyses of residue 417 in regulating the conformation of membrane-anchored full-length SARS-CoV-2 spike and of the functions of the spike in different conformations.
(A) Protein pull-down assay using recombinant human ACE2 as the bait and cell-surface-anchored full-length SARS-CoV-2 spike as the target. The spike contains either Lys417 (wild-type residue) or Val417 (mutant residue). Top: cell-surface-expressed SARS-CoV-2 spike. Middle: pull-down results using His6-tagged ACE2. Bottom: pull-down results using Fc-tagged ACE2 (Figure 3—source data 1). The expected molecular weights of SARS-CoV-2 spike monomer and S2 monomer are ~180 kDa and ~80 kDa, respectively. (B) Flow cytometry assay to detect the interactions between recombinant human ACE2 and cell-surface-anchored full-length SARS-CoV-2 spike (Figure 3—source data 2). The spike contains either Lys417 (wild-type residue) or Val417 (mutant residue) and contains either intact furin motif or inactivated furin motif. See Figure 3—figure supplement 1 for details of this experiment. Data are mean + SEM. A comparison (two-tailed Student’s t-test) was performed on data between indicated groups (n = 3). ***p<0.001. (C) SARS-CoV-2 pseudovirus entry into human-ACE2-expressing cells. The virus-surface-anchored spike contains either Lys417 (wild-type residue) or Val417 (mutant residue). Top: pseudovirus entry efficiency normalized against the expression level of the spike (see bottom) (Figure 3—source data 3). Bottom: SARS-CoV-2 spike in packaged pseudoviruses (Figure 3—source data 4). Data are mean + SEM. A comparison (two-tailed Student’s t-test) was performed on data between indicated groups (n = 8). ***p<0.001. All experiments in this figure were repeated independently three times with similar results.
-
Figure 3—source data 1
Raw image for Figure 3A.
- https://cdn.elifesciences.org/articles/74060/elife-74060-fig3-data1-v3.pptx
-
Figure 3—source data 2
Numerical data for Figure 3B.
- https://cdn.elifesciences.org/articles/74060/elife-74060-fig3-data2-v3.xlsx
-
Figure 3—source data 3
Numerical data for Figure 3C.
- https://cdn.elifesciences.org/articles/74060/elife-74060-fig3-data3-v3.pptx
-
Figure 3—source data 4
Raw image for Figure 3C.
- https://cdn.elifesciences.org/articles/74060/elife-74060-fig3-data4-v3.xlsx
To understand the role of the spike’s conformations in viral entry, we conducted a pseudovirus entry assay. Specifically, retroviruses pseudotyped with SARS-CoV-2 spike (i.e., SARS-CoV-2 pseudoviruses) were evaluated for their capability to enter ACE2-expressing human cells. Two types of SARS-CoV-2 pseudoviruses were used: the K417-spike-charged pseudoviruses (i.e., K417-pseudoviruses) and V417-spike-charged pseudoviruses (V417-pseudoviruses). Both types of pseudoviruses contain the furin motif in their spike. Western blot analysis showed that some of the spike molecules had been cleaved at the S1/S2 boundary when expressed on pseudovirus surface, confirming the intact furin motif (Figure 3C). The pseudovirus entry result showed that V417-pseudoviruses entered cells more efficiently than did K417-pseudoviruses (Figure 3C). Thus, the open spike enhances not only ACE2 binding, but also viral entry.
To understand the role of the spike’s conformation in immune evasion, we analyzed how neutralizing antibodies recognize the RBD in the context of the trimeric spike protein. To this end, we summarized the binding sites of known neutralizing antibodies on the RBD (Figure 4A). These neutralizing antibodies were discovered individually from COVID-19 patients. They generally recognize five groups of neutralizing epitopes on the RBD (Starr et al., 2021; Dussupt et al., 2021; Scheid et al., 2021; Fedry et al., 2021). Three groups of these RBD epitopes are only accessible to neutralizing antibodies when the spike adopts the open conformation (Starr et al., 2021; Dussupt et al., 2021). In comparison, the other two groups of RBD epitopes are accessible to neutralizing antibodies when the spike adopts either open or closed conformation (Scheid et al., 2021; Fedry et al., 2021). In addition to the above conventional antibodies, neutralizing nanobodies (which are single-domain antibodies derived from camelid animals) have also been discovered to target the RBD. Because of their single-domain structure, nanobodies are known to bind to cryptic epitopes on viral targets. We recently discovered four RBD-targeting nanobodies, named Nanosota-1, -2, -3, and -4 (Ye et al., 2021; Ye et al., 2023; Figure 4B). Among them, Nanosota-2 binds to an epitope on the RBD that almost completely overlaps with the ACE2-binding site. Interestingly, the Nanosota-2 epitope is only accessible to Nanosota-2 when the RBD is in the open conformation (Ye et al., 2023). This result suggests that even with the small size of nanobodies, SARS-CoV-2 may be able to evade some of them when its spike is in the closed conformation. Overall, these epitope-based analyses reveal that SARS-CoV-2 spike in the closed conformation can evade significant amounts of neutralizing antibodies, confirming our previous hypothesis that the closed conformation of the spike is a viral strategy for immune evasion (Shang et al., 2020b).
Figure 4

Analyses of epitopes on SARS-CoV-2 receptor-binding domain (RBD) that are accessible to neutralizing antibodies or nanobodies when the spike adopts different conformations.
(A) Epitopes targeted by neutralizing conventional antibodies. Numerous RBD-targeting human antibodies have been discovered individually from COVID-19 patients. They bind to five groups of epitopes. Only one representative antibody for each group is shown. The PDB codes are 7N4L for group 1, 7M7W for group 2, 7M6G for group 3, 7AKD for group 4, and 7M7W for group 5. Three epitopes out of the five groups are accessible to conventional antibodies only when the spike adopts the open conformation. (B) Epitopes targeted by neutralizing nanobodies (single-domain antibodies). We previously discovered four RBD-targeting nanobodies from camelid animals. They bind to three groups of epitopes. The PBD codes are 7KM5 for Nanosota-1, 8G72 for Nanosota-2, 8G74 for Nanosota-3, and 8G75 for Nanosota-4. The Nanosota-2 epitope is accessible to the nanobody only when the spike adopts the open conformation.
To further understand the role of the K417 mutation in ACE2 binding and viral entry, we examined how the K417 mutation directly affects the RBD’s binding affinity for ACE2 instead of the trimeric spike’s. The structure of SARS-CoV-2 RBD complexed with human ACE2 showed that Lys417 in SARS-CoV-2 RBD is directly involved in ACE2 binding by forming a salt bridge with Asp30 in human ACE2 (Lan et al., 2020; Figure 5A). In contrast, the corresponding residue in SARS-CoV-1 RBD is a valine that does not form any direct interactions with human ACE2 (Li et al., 2005; Figure 5B). Here, we introduced the K417V mutation to SARS-CoV-2 RBD and investigated the binding interactions between the recombinant SARS-CoV-2 RBD (containing either Lys417 or Val417) and cell-surface-anchored human ACE2 using flow cytometry. The result showed that the K417V mutation reduced the RBD’s binding affinity for ACE2 (Figure 5C, Figure 5—figure supplement 1). Thus, the K417V mutation has contrasting effects on the RBD and the trimeric spike in terms of ACE2 binding: it weakens the RBD’s ability to bind to ACE2 by eliminating a favorable interaction at the RBD/ACE2 interface, but it increases the trimeric spike’s probability of binding to ACE2 by enabling more spike molecules to assume the open conformation.
Figure 5 with 1 supplement see all
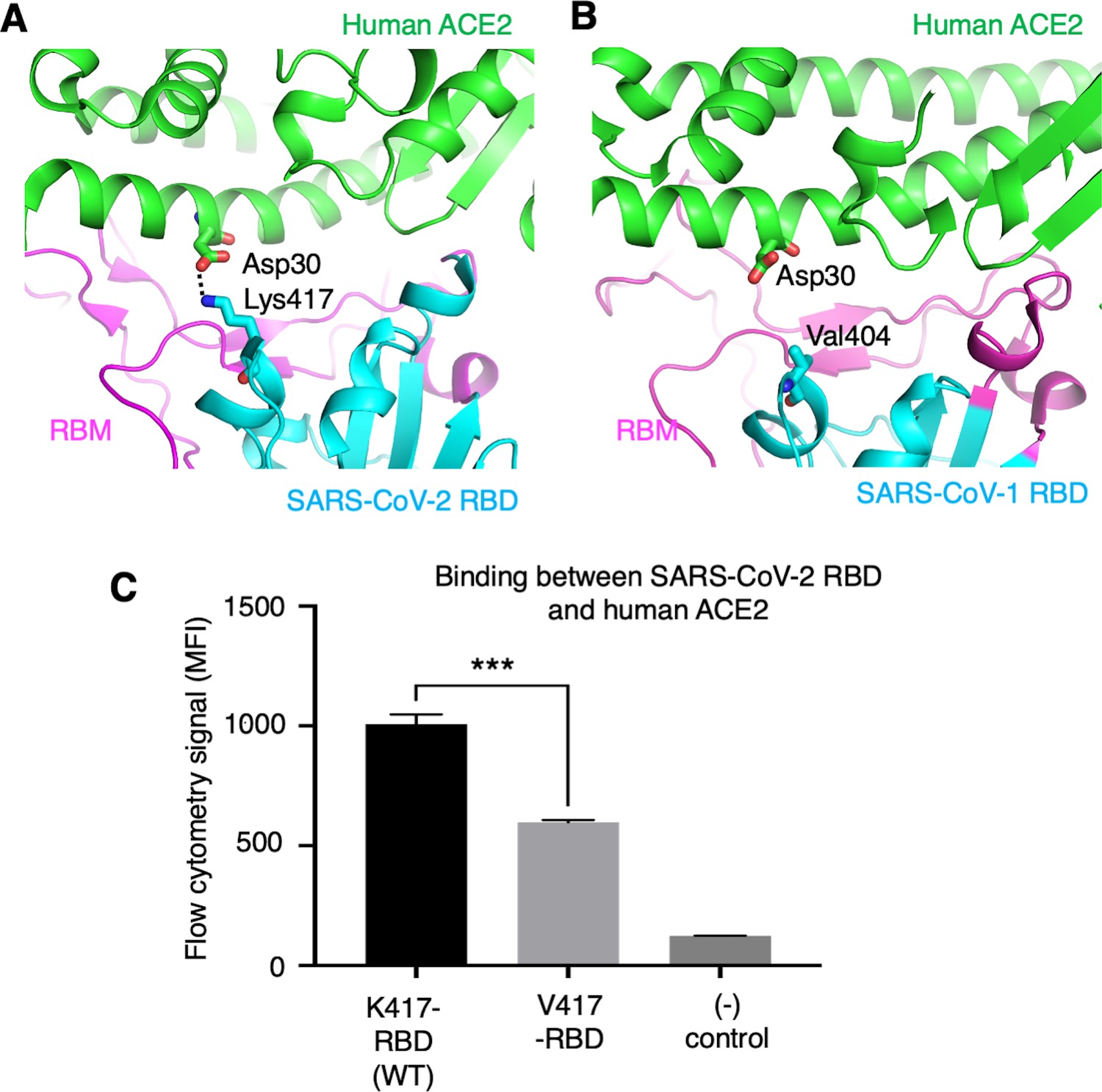
Role of residue 417 in direct interaction with ACE2 receptor.
(A) Lys417 in SARS-CoV-2 receptor-binding domain (RBD) forms a favorable salt bridge with Asp30 in human ACE2. PDB code: 6M0J. (B) Val404 in SARS-CoV-1 RBD (whose position is equivalent to residue 417 in SARS-CoV-2 RBD) does not form any direct interaction with human ACE2. PDB code: 2AJF. (C) Flow cytometry assay to detect the interactions between recombinant SARS-CoV-2 RBD and cell-surface-anchored human ACE2 (Figure 5—source data 1). The RBD contains either Lys417 (wild-type residue) or Val417 (mutant residue). MFI: median fluorescence intensity. See Figure 5—figure supplement 1 for details of this experiment. Data are mean + SEM. A comparison (two-tailed Student’s t-test) was performed on data between indicated groups (n = 3). ***p<0.001. This experiment was repeated independently three times with similar results.
-
Figure 5—source data 1
Numerical data for Figure 5C.
- https://cdn.elifesciences.org/articles/74060/elife-74060-fig5-data1-v3.zip
To summarize, we identified and investigated a molecular switch regulating the conformation of SARS-CoV-2 spike protein. We used multiple approaches: cryo-EM study of recombinant spike ectodomains, biochemical investigation of cell-surface-anchored full-length and wild-type spikes, spike-mediated pseudovirus entry, and structural analysis of neutralizing antibody/nanobody epitopes. To date, several other studies also investigated the conformation of SARS-CoV-2 spike using cryo-EM (Wrobel et al., 2020; Walls et al., 2020; Ke et al., 2020; Xiong et al., 2020; Cai et al., 2020; Benton et al., 2021), some of which gave different ratios of open and closed spikes probably due to differences in sample preparations and/or protein constructions. In this study, the two spike constructs only differ at residue 417 and the two spike samples were prepared using the same procedure. Importantly, our cryo-EM analysis is consistent with our extensive biochemical and functional approaches. These varied experimental methods complement one another, making this study one of the most thorough in examining the conformation of SARS-CoV-2 spike.
Discussion
We previously described a unique feature of the prototypic SARS-CoV-2 spike protein: it is present as a mixture of open and closed conformations (Shang et al., 2020b). Building on this discovery, this study demonstrates that the conformation of SARS-CoV-2 spike is regulated, at least in part, by a structural switch. Additionally, this research sheds light on the roles of the conformational changes in the spike’s functions. These findings have implications for the structure, function, and evolution of coronavirus spikes, as well as for the present and potential future coronavirus infections.
How has SARS-CoV-2 spike evolved to reach a balance of open and closed conformations? Through comparative studies of SARS-CoV-2 and SARS-CoV-1 spikes, we identified a molecular switch, RBD residue 417, that regulates the conformational changes of SARS-CoV-2 spike. Lys417 in the closed SARS-CoV-2 spike forms a hydrogen bond with another spike subunit, stabilizing the closed spike. In contrast, Val417 in SARS-CoV-1 spike cannot form such a hydrogen bond with another spike subunit, unable to stabilize the closed spike. When we introduced the K417V mutation to SARS-CoV-2 spike, more spike molecules turned open. Interestingly, Lys417 in the RBD of SARS-CoV-2 spike also directly interacts with ACE2, and we showed that the K417V mutation reduced the RBD’s binding affinity for ACE2. Hence, the K417V mutation has opposing effects on the RBD and the spike for their binding to ACE2. The net outcomes of the K417 mutation are to enhance the spike’s overall binding to ACE2 and the spike’s capability to mediate viral entry.
Besides residue 417, there are likely other molecular factors that influence the opening and closing of SARS-CoV-2 spike. Studies have shown that N-linked glycans on the spike and fatty acids bound to the spike both play a role in regulating its conformation (Toelzer et al., 2020; Sztain et al., 2021). As for protein-based factors, a D614G mutation that emerged later in the pandemic caused SARS-CoV-2 spike to favor the open conformation (Yurkovetskiy et al., 2020; Ozono et al., 2021). Furthermore, our earlier research indicated that three lysine residues kept SARS-CoV-2 Omicron spike in the open conformation (Ye et al., 2022). It is worth noting that residue 417 itself has been a mutational hotspot in SARS-CoV-2 spike; in some pre-Omicron variants, it changed to either asparagine or threonine, and in most Omicron subvariants, it became asparagine (Figure 1C). Given that the side chains of Asn417 and Thr417 might be too short to form a direct interaction with another spike subunit, the mutations at residue 417 could have influenced the conformation of the spike in subsequent SARS-CoV-2 variants. Nevertheless, this study focuses on prototypic SARS-CoV-2, offering comprehensive structural, biochemical, and functional data that pinpoint residue 417 as a key determinant in regulating the conformation of the prototypic spike. The molecular switch at residue 417 could have been a factor in the onset of the COVID-19 pandemic, as discussed below.
How does the conformation of SARS-CoV-2 spike influence both viral entry and immune evasiveness? Our study shows that, compared to its closed form, the open spike is more effective at binding to ACE2 and facilitating viral entry. This could enhance SARS-CoV-2’s ability to infect and spread among humans. We also found that certain epitopes on the RBD are accessible only in the open spike, allowing them to be targeted by neutralizing conventional antibodies and even compact single-domain nanobodies. This suggests the virus can efficiently evade hosts' immune defenses. The virus' ability to switch between open and closed spike conformations may be key to its balance of infectiousness and evasion of immunity. This delicate balance might explain the often mild symptoms and subdued, delayed immune reactions observed in SARS-CoV-2 carriers. In comparison, SARS-CoV-1 spike predominantly assumes the open conformation (89% open and 11% closed) (Gui et al., 2017), potentially leading to more pronounced symptoms and quicker, stronger immune reactions in those infected. Conversely, NL63-CoV spike remains closed (100% closed) (Walls et al., 2016), possibly resulting in even milder symptoms and more muted immune responses. Therefore, SARS-CoV-2’s blend of infectiousness and evasive abilities, differing from both the highly pathogenic SARS-CoV-1 and the milder NL63-CoV, might be a factor in its widespread transmission.
To summarize, Lys417 in SARS-CoV-2 spike aids in keeping the spike closed, potentially hindering ACE2 binding and viral entry, but providing an edge in immune evasion. Simultaneously, Lys417’s direct interaction with human ACE2 boosts ACE2 binding and viral entry, counteracting the limitations of the closed spike. This interesting mechanism helps SARS-CoV-2 maintain a delicate balance between infectiousness and evading host immune response.
Materials and methods
Cell lines and plasmids
Request a detailed protocolHEK293T cells (American Type Culture Collection [ATCC]) were grown in Dulbecco’s modified Eagle medium (containing 10% fetal bovine serum, 2 mM l-glutamine, 100 units/mL penicillin, and 100 µg/mL streptomycin). 293F cells (Thermo Fisher) were grown in FreeStyle 293 Expression Medium (Thermo Fisher). Both mammalian cells were authenticated by ATCC using STR profiling and were also tested for mycoplasma contamination. No commonly misidentified cell lines were used.
All of the protein constructs in this study were cloned into pcDNA 3.1(+) vector (Life Technologies). Prototypic SARS-CoV-2 spike (GenBank accession number QHD43416.1) and human ACE2 (GenBank accession number NM_021804) were synthesized (GenScript Biotech) and cloned into the vector containing a C-terminal C9 tag. Prototypic SARS-CoV-2 spike ectodomain (residues 1–1211) was cloned into the vector containing either Lys417 or Val417, in addition to six mutations in S2 (F817P, A892P, A899P, A942P, K986P, V987P) (Hsieh et al., 2020; Ye et al., 2022), furin motif mutations (R682A, R683G, R685G) (Shang et al., 2020b), a C-terminal foldon trimerization tag, and a C-terminal His6-tag. SARS-CoV-2 spike RBD (residues 319–535) was cloned into the vector containing either Lys417 or Val417, an N-terminal tPA signal peptide, and a C-terminal His6-tag. Human ACE2 ectodomain (residues 1–615) was cloned into the vector containing either a C-terminal His6-tag or Fc-tag.
Comparison of residue 417 from representative SARS-CoV-2 variants
Request a detailed protocolThe spike sequences of representative SARS-CoV-2 variants were retrieved from https://www.who.int/activities/tracking-SARS-CoV-2-variants.
Protein expression and purification
Request a detailed protocolAll of the recombinant proteins were expressed in 293F cells (Thermo Fisher) using a FreeStyle 293 mammalian cell expression system (Life Technologies) as previously described (Wan et al., 2020b). In brief, the His-tagged proteins were collected from cell culture medium, purified using a Ni-NTA column (Cytiva Healthcare), and purified further using a Superdex gel filtration column (Cytiva Healthcare). The Fc-tagged protein was purified in the same way as the His-tagged proteins, except that a protein A column replaced the Ni-NTA column in the procedure.
Protein pull-down assay
Request a detailed protocolProtein pull-down assay was performed using a Dynabeads immunoprecipitation kit (Invitrogen) as previously described (Shang et al., 2020b). Briefly, 10 μL of Dynabeads, either for His6-tagged proteins or for Fc-tagged proteins, were washed with PBS buffer and then were incubated with either 8 μg ACE2-His (human ACE2 with a C-terminal His6 tag) or 10 μg ACE2-Fc (human ACE2 with a C-terminal Fc tag). Subsequently, ACE2-bound beads were washed with PBS buffer plus 0.05% Tween-20 (PBST) and then were aliquoted into different tubes for later use. To prepare cell-associated SARS-CoV-2 spike (containing either Lys417 or Val417), HEK293T cells were transfected with pcDNA3.1(+) plasmid encoding the full-length spike (containing a C-terminal C9 tag). 48 hr after transfection, the spike-expressing cells were lysed in immunoprecipitation assay buffer using a sonicator and then centrifuged. The supernatants containing the solubilized cell-membrane-associated spike were incubated with the ACE2-bound beads (ACE2 was in excess of spike). The beads were then washed with PBST buffer, and the bound spike was eluted using elution buffer. The samples were then subjected to western blot analysis and detected using anti-C9-tag antibody (Thermo Fisher).
Flow cytometry
Request a detailed protocolFlow cytometry was performed to detect the interactions between recombinant human ACE2 and cell-surface-expressed full-length SARS-CoV-2 spike (containing either Lys417 or Val417 and containing either intact furin motif or inactivated furin motif). The procedure was carried out as previously described (Maeda et al., 2022; Zhang et al., 2020). Briefly, the plasmids encoding one of the C9-tagged SARS-CoV-2 spikes or vector (pcDNA3.1(+)) itself were transfected into HEK293T cells. After 36 hr, the cells were harvested and incubated with recombinant Fc-tagged human ACE2 (25 µg/mL) on ice for 1 hr. After three washes with PBS (containing 1% BSA), the cells were incubated with Alexa Fluor 488 anti-human-IgG-Fc antibody (1:500) (BioLegend Inc) and PE-labeled anti-C9-tag antibody (1:500) (Santa Cruz Biotechnology) on ice for another hour. After more washes, the cells were fixed with 4% formaldehyde. The fluorescence intensities of the cells were measured using flow cytometry (BD FACSymphony A3 cell analyzer).
Flow cytometry was also performed to detect the interactions between SARS-CoV-2 RBD (containing either Lys417 or Val417) and cell-surface-expressed human ACE2. Briefly, HEK293T cells stably expressing human ACE2 were constructed as previously described (Shang et al., 2018; Geng et al., 2021). These cells were incubated with one of the recombinant His-tagged SARS-CoV-2 RBDs (10 µg/mL) or buffer only on ice for 1 hr. After three washes with PBS (containing 1% BSA), the cells were incubated with PE-labeled anti-His-tag antibody (1:500) (Santa Cruz Biotechnology) on ice for another hour. After more washes, the cells were fixed with 4% formaldehyde, and the fluorescence intensity of the cells was measured using flow cytometry.
Pseudovirus entry
Request a detailed protocolPseudoviruses were packaged as previously described (Peng et al., 2017). Briefly, pcDNA3.1(+) plasmid encoding the full-length SARS-CoV-2 spike (containing either Lys417 or Val417) was co-transfected into HEK293T cells with helper plasmid psPAX2 and reporter plasmid plenti-CMV-luc at a molar ratio of 1:1:1 using Lipofectamine 3000 (Life Technologies). The produced pseudoviruses were harvested 72 hr post transfection and then were used to enter HEK293T cells stably expressing human ACE2. After incubation at 37°C for 5 hr, medium was replaced and cells were incubated for an additional 48 hr. Cells were then washed with PBS and lysed. Aliquots of cell lysates were transferred to Optiplate-96 (PerkinElmer), followed by addition of luciferase substrate. Relative light units (RLUs) were measured using EnSpire plate reader (PerkinElmer). Meanwhile, the amounts of pseudovirus-packaged spikes were measured by western blot using anti-C9-tag antibody and then quantified using Fiji (https://imagej.net/). The RLUs were then normalized against the amounts of pseudovirus-packaged spikes.
Western blot
Request a detailed protocolCells or pseudoviruses were mixed with SDS loading buffer and then were incubated at 95°C for 10 min. Samples were run in a 10% SDS Tris-Glycine Gel and transferred to a PVDF membrane. Anti-C9-tag or anti-His-tag monoclonal primary antibody (1:1000) (Santa Cruz Biotech) and horseradish peroxidase-conjugated mouse secondary antibody (1:10,000) (Jackson Laboratory) were used for western blotting. LAS-4000 imager (Cytiva Healthcare) was used to develop images.
Cryo-EM grid preparation and data acquisition
Request a detailed protocolThe recombinant spike ectodomain (containing either Lys417 or Val417) (4 µL at 10.3–11.5 μM) was applied to freshly glow-discharged Quantifoil R1.2/1.3 300-mesh copper grids (EM Sciences), and then blotted for 4 s at 22°C under 100% chamber humidity and plunge-frozen in liquid ethane using a Vitrobot Mark IV (FEI). Cryo-EM data were collected using Latitude-S (Gatan) on a Titan Krios electron microscope (Thermo Fisher) equipped with a K3 direct electron detector with a Biocontinuum energy filter (Gatan) in counting mode at the Hormel Institute, University of Minnesota. The movies were collected at a nominal magnification of 130,000× (corresponding to 0.664 Å per pixel), a 20 eV slit width, a dose rate of 20 e- per Å2 per second, and a total dose of 40 e-/Å2. The statistics of cryo-EM data collection are summarized in Supplementary file 1.
Image processing
Request a detailed protocolCryo-EM data were processed using cryoSPARC v4.0.3 (Punjani et al., 2017), and the data processing procedures are outlined in Figure 2—figure supplements 1 and 2. Dose-fractionated movies were first subjected to patch motion correction with data downsampled by 4/3 (0.885333 Å/pixel after downsampling) and patch CTF estimation with MotionCor2 (Rubinstein and Brubaker, 2015) and CTFFIND-4.1.13 (Rohou and Grigorieff, 2015), respectively. Images with the defocus values outside of –0.8 to –2.4 μm or the CTF fit resolutions worse than 6 Å were excluded from further steps. Particles were picked using both Blob picker and Template picker accompanied by removing duplicate particles. Three rounds of 2D classifications were applied to remove junk particles, and good particles (57,795 or 45,999) extracted from the good 2D classes were used for ab initio reconstruction of three maps and then for the heterogeneous refinements. The good 3D class (42,970 or 29,596 particles) was finally subjected to further homogeneous, non-uniform, and CTF refinements to generate a 3.4 Å or 3.7 Å resolution map.
Particles in the good 3D classes were then imported into RELION-4.0 (Zivanov et al., 2020) using the csparc2star.py module (UCSF pyem v0.5. Zenodo) and subjected to signal subtraction to keep only the mixed-state receptor-binding subunit of the spike, followed by masked 3D classification. Particles in the two different classes from the masked 3D classification were then reverted to the original particles and subjected to non-uniform refinements in cryoSPARC v3.3.2. The C3 symmetry was applied for the maps with the closed state RBD. Map resolution was determined by gold-standard Fourier shell correlation (FSC) at 0.143 between the two half-maps. Local resolution variation was estimated from the two half-maps in cryoSPARC v4.0.3.
Model building and refinement
Request a detailed protocolInitial model building of the prototypic SARS-CoV-2 spike was performed in Coot-0.8.9 (Emsley and Cowtan, 2004) using PDBs 7TGX and 7TGY as the starting models. Several rounds of refinement in Phenix-1.16 (Adams et al., 2010) and manually building in Coot-0.8.9 were performed until the final reliable models were obtained. The final model has good stereochemistry as evaluated by MolProbity (Chen et al., 2010). The statistics of 3D reconstruction and model refinement are shown in Supplementary file 1. Figures were generated using UCSF Chimera X v0.93 (Goddard et al., 2018).
In this study, both open spikes have only one RBD standing up and two RBDs lying down. This finding aligns with the results of several other structural studies on SARS-CoV-2 spike (Walls et al., 2020; Xiong et al., 2020; Cai et al., 2020). However, some other studies have reported instances of open SARS-CoV-2 spike with either two or three RBDs standing up (Benton et al., 2021; Yurkovetskiy et al., 2020). The cause for this discrepancy is unclear, but could be due to different sample preparations and/or protein constructions.
Data availability
All data generated or analysed during this study are included in the manuscript and supporting files. Source data files have been provided for Figures 3 and 5. The atomic models and corresponding cryo-EM density maps have been deposited into the PDB and the Electron Microscopy Data Bank, respectively, with accession numbers PDB 8UUL and EMD-42589 (prototypic SARS-CoV-2 spike containing Lys417 in the closed conformation), PDB 8UUM and EMD-42590 (prototypic SARS-CoV-2 spike containing Lys417 in the open conformation), PDB 8UUN and EMD-42591 (prototypic SARS-CoV-2 spike containing Val417 in the closed conformation), and PDB 8UUO and EMD-42592 (prototypic SARS-CoV-2 spike containing Val417 in the open conformation).
-
RCSB Protein Data BankID 8UUL. Prototypic SARS-CoV-2 spike containing Lys417 in the closed conformation.
-
Electron Microscopy Data BankID EMD-42589. Prototypic SARS-CoV-2 spike containing Lys417 in the closed conformation.
-
RCSB Protein Data BankID 8UUM. Prototypic SARS-CoV-2 spike containing Lys417 in the open conformation.
-
Electron Microscopy Data BankID EMD-42590. Prototypic SARS-CoV-2 spike containing Lys417 in the open conformation.
-
RCSB Protein Data BankID 8UUN. Prototypic SARS-CoV-2 spike containing Val417 in the closed conformation.
-
Electron Microscopy Data BankID EMD-42591. Prototypic SARS-CoV-2 spike containing Val417 in the closed conformation.
-
RCSB Protein Data BankID 8UUO. Prototypic SARS-CoV-2 spike containing Val417 in the open conformation.
-
Electron Microscopy Data BankID EMD-42592. Prototypic SARS-CoV-2 spike containing Val417 in the open conformation.
References
-
PHENIX: a comprehensive Python-based system for macromolecular structure solutionActa Crystallographica. Section D, Biological Crystallography 66:213–221.https://doi.org/10.1107/S0907444909052925
-
MolProbity: all-atom structure validation for macromolecular crystallographyActa Crystallographica. Section D, Biological Crystallography 66:12–21.https://doi.org/10.1107/S0907444909042073
-
The spike protein of SARS-CoV--a target for vaccine and therapeutic developmentNature Reviews. Microbiology 7:226–236.https://doi.org/10.1038/nrmicro2090
-
Coot: model-building tools for molecular graphicsActa Crystallographica. Section D, Biological Crystallography 60:2126–2132.https://doi.org/10.1107/S0907444904019158
-
Distinctive roles of furin and TMPRSS2 in SARS-CoV-2 infectivityJournal of Virology 96:e0012822.https://doi.org/10.1128/jvi.00128-22
-
A study on infectivity of asymptomatic SARS-CoV-2 carriersRespiratory Medicine 169:106026.https://doi.org/10.1016/j.rmed.2020.106026
-
Structural basis for human receptor recognition by SARS-CoV-2 Omicron Variant BA.1Journal of Virology 96:e0024922.https://doi.org/10.1128/jvi.00249-22
-
Asymptomatic patients as A source of COVID-19 infections: a systematic review and meta-analysisInternational Journal of Infectious Diseases 98:180–186.https://doi.org/10.1016/j.ijid.2020.06.052
-
A major outbreak of severe acute respiratory syndrome in Hong KongThe New England Journal of Medicine 348:1986–1994.https://doi.org/10.1056/NEJMoa030685
-
Receptor recognition mechanisms of coronaviruses: a decade of structural studiesJournal of Virology 89:1954–1964.https://doi.org/10.1128/JVI.02615-14
-
Structure, function, and evolution of coronavirus spike proteinsAnnual Review of Virology 3:237–261.https://doi.org/10.1146/annurev-virology-110615-042301
-
Early transmission dynamics in Wuhan, China, of novel coronavirus-infected pneumoniaThe New England Journal of Medicine 382:1199–1207.https://doi.org/10.1056/NEJMoa2001316
-
Structural and molecular evidence suggesting coronavirus-driven evolution of mouse receptorThe Journal of Biological Chemistry 292:2174–2181.https://doi.org/10.1074/jbc.M116.764266
-
CTFFIND4: Fast and accurate defocus estimation from electron micrographsJournal of Structural Biology 192:216–221.https://doi.org/10.1016/j.jsb.2015.08.008
-
Alignment of cryo-EM movies of individual particles by optimization of image translationsJournal of Structural Biology 192:188–195.https://doi.org/10.1016/j.jsb.2015.08.007
-
A glycan gate controls opening of the SARS-CoV-2 spike proteinNature Chemistry 13:963–968.https://doi.org/10.1038/s41557-021-00758-3
-
Identification of a new human coronavirusNature Medicine 10:368–373.https://doi.org/10.1038/nm1024
-
Glycan shield and epitope masking of a coronavirus spike protein observed by cryo-electron microscopyNature Structural & Molecular Biology 23:899–905.https://doi.org/10.1038/nsmb.3293
-
Molecular mechanism for antibody-dependent enhancement of coronavirus entryJournal of Virology 94:e02015-19.https://doi.org/10.1128/JVI.02015-19
-
Author Correction: SARS-CoV-2 and bat RaTG13 spike glycoprotein structures inform on virus evolution and furin-cleavage effectsNature Structural & Molecular Biology 27:1001.https://doi.org/10.1038/s41594-020-0509-2
-
A thermostable, closed SARS-CoV-2 spike protein trimerNature Structural & Molecular Biology 27:934–941.https://doi.org/10.1038/s41594-020-0478-5
-
Cryo-EM structure of a SARS-CoV-2 omicron spike protein ectodomainNature Communications 13:1214.https://doi.org/10.1038/s41467-022-28882-9
-
Structural evolution of SARS-CoV-2 omicron in human receptor recognitionJournal of Virology 97:e0082223.https://doi.org/10.1128/jvi.00822-23
Decision letter
-
Lejla ZubcevicReviewing Editor; University of Kansas Medical Center, United States
-
Diane M HarperSenior Editor; University of Michigan, United States
-
Lejla ZubcevicReviewer; University of Kansas Medical Center, United States
-
Yi RenReviewer; Vanderbilt University, United States
In the interests of transparency, eLife publishes the most substantive revision requests and the accompanying author responses.
Decision letter after peer review:
Thank you for submitting your article "Molecular switches regulating the potency and immune evasiveness of SARS-CoV-2 spike protein" for consideration by eLife. Your article has been reviewed by 3 peer reviewers, including Lejla Zubcevic as Reviewing Editor and Reviewer #1, and the evaluation has been overseen by José Faraldo-Gómez as Senior Editor. The following individual involved in review of your submission has agreed to reveal their identity: Yi Ren (Reviewer #3).
The reviewers have discussed their reviews with one another, and the Reviewing Editor has drafted this to help you prepare a revised submission.
Essential revisions:
All reviewers agreed that this is a well-executed study with significant findings. However, we agreed that a few revisions should be made prior to publication. We have compiled the list of requests, comments, and questions below.
1. In order to be able to comment on the molecular mechanism of close-to-open transition, the authors need to look at atomic models of the RBD captured in both closed and open states. The reviewers would like the authors to submit an open RBD structure. The data for this should already exist (4.6A K417V/furin motif deletion mutant 3D reconstruction presented in figure 2C). To get an idea of the general mechanism of the closed-to-open transition, the authors could build a (where appropriate, side chain-less model) of the 4.6A K417V/furin motif deletion mutant, for which the data already exists.
2. We would encourage both a comparison with the FnM-deletion (100% closed) structure and an internal comparison between the open and closed protomers in the "open" structure to gain an insight into the mechanism.
3. The way it is explained in the methods, it would appear that only the highest resolution class is taken into consideration when the percentage occupancies are calculated for the open and closed states. We are not convinced that this is the most appropriate way to analyze the data because it is inherently biased towards more structurally stable classes. Other open conformations might exist at lower resolutions. Ideally, the state occupancy percentages should be based on the total particle count. For example, assuming that the open conformation is more unstable than the closed one, the real distribution could be 90% open and 10% closed, where only the 10% are selected and contribute to the high resolution reconstruction. In this case, it'd be wrong to say that 100% of the particles are in the closed conformation when the data actually says that 100% of the high resolution particles (and 10% of the total) are in the closed conformation. At the very least, the authors should discuss this aspect of data analysis and explain the implications for their hypothesis.
4. The authors should create a figure that explains their cryo-EM workflow for analysis in detail (representative micrograph, 2D classification, 3D classifications, etc). This figure should also contain local resolution plots and Euler angle distribution plots.
5. The PDB validation and visual inspection of maps indicates that lots of residues/regions do not fit the map very well. Lots of side chains were built with no density supporting them. We encourage the authors to go back to these models and a. remove segments of the model that are not supported by experimental density and b. re-build the backbone of the protein in parts of the protein where it does not fit the density well. This is particularly an issue because the authors discuss interaction networks in regions with poor density (Lys417 and it's interaction partner are part of this region too). As requested in point 4, a local resolution plot of the 3D model will also come in handy for the readers here to easily estimate the accuracy of the map in different regions of the protein.
6. The authors do not show a WT control in their mouse immunization experiments (figure 5). This control should be included.
7. The authors should comment on why we only observe one RBD in the up conformation, despite point mutations, especially Lys417Ala, being introduced to all 3 protomers? Are there factors that might be missing from the experimental system that might play a role in stabilizing the "fully open" i.e. all 3 RBDs in the up conformation.
8. Is there co-operativity in the trimer? E.g., does releasing one RBD change the open-to-closed equilibrium for the other two? And do the furin motif and the salt bridge act independently of each other?
9. The WT SARS-CoV-2 (containing the FnM) still performs better than the point mutant (FnM-point) in virus entry assays, suggesting that the charges within the FnM might play an important role. Have the authors created a SARS-CoV-2 with a GSGS-linker for comparison? And SARS-CoV-2 with the Arg replaced with Lys (which wouldn't be cleaved by furin)?
10. Is it accurate that the structure of RaTG13-CoV is always in the closed conformation or is this an artefact of experimental conditions? This structure was crosslinked, which could have led to a 100% closed population.
11. Is it known what allows the RaTG13 spike molecule to switch into an open conformation? Are charged residues expected at play in this scenario as well (i.e., does RaTG13 have a lysine residue in the same position as SARS-CoV-2)? For comparison purposes, it would be helpful to know what residue is at the Lys417-equivalent position in RaTG13.
12. How well does the WT SARS-CoV-1 spike construct (controlled for pre-fusion) bind ACE2 and behave in the pseudovirus entry experiment? Is it comparable to the SARS-CoV-2 K417V mutant? If it is, this would suggest that the two major switches for dictating the open/closed confirmations are predominantly due to the presence of the furin motif loop and the salt bridge. If not, this could point to other factors involved in contributing to the conformational differences between SARS-CoV-1 and -2.
13. Lys417 has been identified as important for ACE2 binding. Can the authors comment on this, in the light of this new data.
14. Glycans and fatty acids have also been suggested to play a role in the open-to-closed transition. Can the authors comment on their potential roles in the light of this new data.
15. The illustration of the mechanism of the closed-to-open transition Figure S3A was difficult to interpret. Similarly, the interfaces between S1 and NTD were also difficult to glean from the figure. We would encourage the authors to make two separate figures (or at least, two panels in one figure) to illustrate this better. Also, a surface representation might work better to show the interface between S1 and NTD.
16. Could the authors include more spike structures (i.e. closed form of D614G) for comparing the interfaces in Figure S3? Spike proteins features extensive conformational heterogeneity. Even within the same category of open or closed spike, further classification generates slightly different structures. It is not entirely clear what level of changes is significant.
17. Line 99-101: The authors should quote A. C. Walls et al., Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell, (2020).
18. Line 136-137: Reference for the double proline/C-term foldon.
19. It would be helpful to include expected molecular masses in the legends of Figures 1C, 3B and 4B where expected bands should appear.
20. It would be very helpful to include a table of experimental outcomes that includes the protein conformations of all WT proteins (CoV-1, CoV-2, and RaTG13) and mutants, a summary of ACE2 binding and pseudovirus entry.
21. In line 441, is "26,126 particles" correct? This number seems to refer to particles selected from 2D for the entire dataset.
Reviewer #1 (Recommendations for the authors):
The conformation of the SARS-CoV-2 spike protein receptor binding domain (RBD) has implications for binding to the host cell surface receptor: the open conformation is required for binding, but it also makes the virus more likely to be detected by the host's immune system. SARS-CoV-2 spike protein RBD has been observed with equal probability in both open and closed conformations but the mechanism of the transition from the closed to the open state is not known.
Here, Wan et al. present data that suggests that the flexible linker between S1 and S2, which contains a furin binding motif, is an important factor in facilitating this transition.
Importantly, the authors' findings suggest that it is not the furin motif itself but the flexible linker that facilitates the transition to the open conformation.
The study stretches from basic biochemistry to structural biology to in vivo experiments. However, the work can be strengthened by some additional data, data analysis and improved presentation of the structural biology section. In addition, some clarification regarding the in vivo data is required.
The authors present an interesting hypothesis that by being equally able to occupy the open and closed RBD conformations, SARS-Cov-2 has evolved to have high viral potency as well as successfully evade the host immune response. This is an interesting insight into viral evolution which may lead to novel avenues for future research.
I have the following comments:
1. To be able to comment on the molecular mechanism of close-to-open transition, the authors need to look at atomic models of the RBD captured in both closed and open states. The reviewers would like the authors to submit an open RBD structure. The data for this should already exist (4.6A K417V/furin motif deletion mutant 3D reconstruction presented in figure 2C). To get an idea of the general mechanism of the closed-to-open transition, the authors could build a (where appropriate, side chain-less model) of the 4.6A K417V/furin motif deletion mutant, for which the data already exists.
2. The authors should compare the open RBD structure with the FnM-deletion (100% closed) structure. In addition, they should make an internal comparison between the open and closed protomers in the "open" structure to gain an insight into the mechanism.
3. The way it is explained in the methods, only the highest resolution class is taken into consideration when the percentage occupancies are calculated for the open and closed states. This approach is inherently biased towards more structurally stable classes. Other open conformations might exist at lower resolutions. Ideally, the state occupancy percentages should be based on the total particle count.
For example, assuming that the open conformation is more unstable than the closed one, the real distribution could be 90% open and 10% closed, where only the 10% are selected and contribute to the high-resolution reconstruction. In this case, it'd be wrong to say that 100% of the particles are in the closed conformation when the data actually says that 100% of the high-resolution particles (and 10% of the total) are in the closed conformation. At the very least, the authors should discuss this aspect of data analysis and explain the implications for their hypothesis.
4. The authors should create a figure that explains their cryo-EM workflow for analysis in detail (representative micrograph, 2D classification, 3D classifications, etc). This figure should also contain local resolution plots and Euler angle distribution plots.
5. The PDB validation and visual inspection of maps indicates that lots of residues/regions do not fit the map very well. Lots of side chains were built with no density supporting them. The authors should: a. remove segments of the model that are not supported by experimental density and b. re-build the backbone of the protein in parts of the protein where it does not fit the density well. This is particularly an issue because the authors discuss interaction networks in regions with poor density (Lys417 and its interaction partner are part of this region too).
6. The authors do not show a WT control in their mouse immunization experiments (figure 5). This control should be included.
7. The authors should discuss the Lys417 residue and its involvement in ACE2 binding.Reviewer #2 (Recommendations for the authors):
Wan et al., investigate some of the major structural differences between the SARS-CoV-1, SARS-CoV-2, and RaTG13-CoV spike proteins to better understand why SARS-CoV-2 led to a global pandemic in humans. In particular, the study focuses on the SARS-CoV-2 spike (S) protein in the pre-fusion conformation, used by the virus to bind and enter host cells. The pre-fusion conformation of the S protein is known to have either open, closed or a mixture of open/closed conformations within the receptor-binding domain (RBD), depending on the type of coronavirus. The SARS-CoV-2 RBD is found in both the open and closed conformations while SARS-CoV-1 and RaTG13-CoV are found in the open and closed, respectively. The authors investigate the contributions of two unique structural features of the SARS-CoV-2 S protein, a furin binding motif within a loop as well as a lysine residue (position 417), to understand the observed mixture of conformations. Using a combination of mutational analyses, biochemical assays, structural studies, and mouse models, the authors show that both structural motifs (the furin loop and lysine residue) contribute to the ability of the protein to switch between the open and closed conformations. Furthermore, this switching between conformations most likely contributes to the effectiveness of SARS-CoV-2 establishing widespread infections in humans. Overall, this very interesting study is well conducted and will aid in the development of additional SARS-CoV-2 studies, as well as inform how other coronaviruses may employ the same strategies.
Comments:
1) It would be important to know how well the WT SARS-CoV-1 spike construct (controlled for pre-fusion) binds ACE2 and behaves in the pseudovirus entry experiment. Is it comparable to the SARS-CoV-2 K417V mutant? If it is, this would suggest that the two major switches for dictating the open/closed confirmations are predominantly due to the presence of the furin motif loop and the salt bridge. If not, this could point to other factors involved in contributing to the conformational differences between SARS-CoV-1 and -2.Reviewer #3 (Recommendations for the authors):
In this work, the authors have aimed to identify molecular switches regulating the conformations of SARS-CoV-2 spike protein and to investigate their effects on the potency and immune evasiveness of SARS-CoV-2. The authors have identified two such molecular switches, the furin motif and the K417 residue at the interface of the RBD domains in closed spike. The strength of the work is the comprehensive approach (SARS-CoV-2/RaTG13-CoV engineering, ACE2 binding, cryo-EM, pseudovius entry assays, etc). The authors clearly demonstrate the effects of furin motif and K417 on the ratios of open and closed spikes, and consistently, on ACE2 binding and pseudovirus entry. The correlation between spike conformations and immune evasiveness is an intriguing observation, although it remains to be more vigorously tested in future studies.
[Editors’ note: further revisions were suggested prior to acceptance, as described below.]
Thank you for resubmitting your work entitled "Lys417 acts as a molecular switch that regulates the conformation of SARS-CoV-2 spike protein" for further consideration by eLife. Your revised article has been evaluated by Diane Harper (Senior Editor) and a Reviewing Editor.
The manuscript has been improved but there are some remaining issues that need to be addressed, as outlined below:
Reviewer #1 (Recommendations for the authors):
Here, Li et al. identify a molecular switch within the SARS-CoV-1/2 spike proteins, in position 417. Based on comparisons between CoV-1 and CoV-2 sequences, the authors propose that the nature of the residue of this position determines whether the spike favors a closed or an open conformation. The authors present cryo-EM and biochemical data in support of their hypothesis. The paper will be of general interest to those interested in molecular mechanisms of viral infection by SARS-CoV-2 and other coronaviruses.
The authors have submitted a streamlined and improved manuscript. I only have a few comments to add.
– My main concern is that the word "affinity" is not used properly in some parts of the paper. For example, in the abstract, the authors state "The net outcomes of this (K417V) mutation are to allow the spike to bind ACE2 more tightly…". This is not really what the data says- the mutation seems to increase the probability of "open" spike and therefore increases the probability of spike binding. In fact, the data shows that K417V binds less tightly to ACE2 than K417.
In lines 116-118 the authors write "It has been shown that ACE2 only binds to the standing-up RBD in the open spike (28, 33), suggesting that open spike binds to ACE2 with higher affinity than the closed spike." A more accurate statement would be that the open spike conformation is necessary for ACE2 binding.
This continues in lines 127-129 "The pull-down result showed that compared to K417-spike, more V417-spike molecules had been pulled down by ACE2, suggesting that V417-spike has a higher affinity for ACE2 (Figure 3A)." This experiment does not say anything about binding affinity, but it does suggest that the binding probability between the spike protein and ACE2 is increased in V417.
In the summary of findings in lines 184-187: "it weakens the RBD's ability to bind to ACE2 by eliminating a favorable interaction at the RBD/ACE2 interface, but it strengthens the trimeric spike's binding affinity for ACE2 by enabling more spike molecules to assume the open conformation." A more accurate statement would be that the V417 increases the probability of binding to ACE2 because it increases the probability of the open spike conformation.
https://doi.org/10.7554/eLife.74060.sa1Author response
Essential revisions:
All reviewers agreed that this is a well-executed study with significant findings. However, we agreed that a few revisions should be made prior to publication. We have compiled the list of requests, comments, and questions below.
1. In order to be able to comment on the molecular mechanism of close-to-open transition, the authors need to look at atomic models of the RBD captured in both closed and open states. The reviewers would like the authors to submit an open RBD structure. The data for this should already exist (4.6A K417V/furin motif deletion mutant 3D reconstruction presented in figure 2C). To get an idea of the general mechanism of the closed-to-open transition, the authors could build a (where appropriate, side chain-less model) of the 4.6A K417V/furin motif deletion mutant, for which the data already exists.
The revised manuscript centers on residue 417, which acts as a molecular switch that regulates the conformation of SARS-CoV-2 spike. It has omitted the data and discussion about the furin motif deletion mutant (Figure 1). Accordingly, the title of the manuscript has been changed to “Lys417 acts as a molecular switch regulating the conformation of SARS-CoV-2 spike protein”.
In the revised manuscript, we have developed new stabilized versions of SARS-CoV-2 spike ectodomain. This was achieved by incorporating six proline mutations into the S2 subunit, an increase from the two proline mutations used in earlier versions. Subsequently, we acquired new cryo-EM data for these proteins at significantly improved resolutions and constructed new atomic models for each of the samples (Figure 2).
2. We would encourage both a comparison with the FnM-deletion (100% closed) structure and an internal comparison between the open and closed protomers in the "open" structure to gain an insight into the mechanism.
The revised manuscript centers on residue 417, which acts as a molecular switch that regulates the conformation of SARS-CoV-2 spike. It has omitted the data and discussion about the furin motif deletion mutant (Figure 1). Accordingly, the title of the manuscript has been changed to “Lys417 acts as a molecular switch regulating the conformation of SARS-CoV-2 spike protein”.
3. The way it is explained in the methods, it would appear that only the highest resolution class is taken into consideration when the percentage occupancies are calculated for the open and closed states. We are not convinced that this is the most appropriate way to analyze the data because it is inherently biased towards more structurally stable classes. Other open conformations might exist at lower resolutions. Ideally, the state occupancy percentages should be based on the total particle count. For example, assuming that the open conformation is more unstable than the closed one, the real distribution could be 90% open and 10% closed, where only the 10% are selected and contribute to the high resolution reconstruction. In this case, it'd be wrong to say that 100% of the particles are in the closed conformation when the data actually says that 100% of the high resolution particles (and 10% of the total) are in the closed conformation. At the very least, the authors should discuss this aspect of data analysis and explain the implications for their hypothesis.
Please see Figure 2—figure supplement1, Figure 2—figure supplement2, and Supplementary File 1 for the updated cryo-EM procedures.
4. The authors should create a figure that explains their cryo-EM workflow for analysis in detail (representative micrograph, 2D classification, 3D classifications, etc). This figure should also contain local resolution plots and Euler angle distribution plots.
Please see Figure 2—figure supplement1 and Figure 2—figure supplement2 for the cryo-EM workflow.
5. The PDB validation and visual inspection of maps indicates that lots of residues/regions do not fit the map very well. Lots of side chains were built with no density supporting them. We encourage the authors to go back to these models and a. remove segments of the model that are not supported by experimental density and b. re-build the backbone of the protein in parts of the protein where it does not fit the density well. This is particularly an issue because the authors discuss interaction networks in regions with poor density (Lys417 and it's interaction partner are part of this region too). As requested in point 4, a local resolution plot of the 3D model will also come in handy for the readers here to easily estimate the accuracy of the map in different regions of the protein.
The cryo-EM densities and the corresponding atomic models presented in this revision represent a substantial improvement over our previous submission. For detailed comparisons and data, refer to Figure 2—figure supplement1, Figure 2—figure supplement2, and Supplementary File 1.
6. The authors do not show a WT control in their mouse immunization experiments (figure 5). This control should be included.
The revised manuscript no longer includes mouse immunization data. Instead, it presents findings that the closed spike can evade the binding of many neutralizing antibodies and nanobodies (Figure 4).
7. The authors should comment on why we only observe one RBD in the up conformation, despite point mutations, especially Lys417Ala, being introduced to all 3 protomers? Are there factors that might be missing from the experimental system that might play a role in stabilizing the "fully open" i.e. all 3 RBDs in the up conformation.
We have added the following comment to the revised manuscript:
“In this study, both of the open spikes have only one RBD standing up and two RBDs lying down. This finding aligns with the results of several other structural studies on SARS-CoV-2 spike (29, 40, 41). However, some other studies have reported instances of open SARS-CoV-2 spike with either two or three RBDs standing up (42, 45). The cause for this discrepancy is unclear, but could be due to different sample preparations and/or protein constructions.”
8. Is there co-operativity in the trimer? E.g., does releasing one RBD change the open-to-closed equilibrium for the other two? And do the furin motif and the salt bridge act independently of each other?
We don't have the answers to the first two questions, and we believe they fall outside the scope of our current study. Regarding the last question, we have excluded data and discussions about the furin deletion mutant spike from the revised manuscript. Instead, the focus has shifted to residue 417 in the revised manuscript, pinpointing it as an important determinant that regulates the conformation of SARS-CoV-2 spike.
9. The WT SARS-CoV-2 (containing the FnM) still performs better than the point mutant (FnM-point) in virus entry assays, suggesting that the charges within the FnM might play an important role. Have the authors created a SARS-CoV-2 with a GSGS-linker for comparison? And SARS-CoV-2 with the Arg replaced with Lys (which wouldn't be cleaved by furin)?
We have excluded data and discussions about the furin deletion mutant spike from the revised manuscript. Instead, the focus has shifted to residue 417 in the revised manuscript, pinpointing it as an important determinant that regulates the conformation of SARS-CoV-2 spike.
10. Is it accurate that the structure of RaTG13-CoV is always in the closed conformation or is this an artefact of experimental conditions? This structure was crosslinked, which could have led to a 100% closed population.
The revised manuscript no longer includes discussion regarding the RaTG13-CoV spike.
11. Is it known what allows the RaTG13 spike molecule to switch into an open conformation? Are charged residues expected at play in this scenario as well (i.e., does RaTG13 have a lysine residue in the same position as SARS-CoV-2)? For comparison purposes, it would be helpful to know what residue is at the Lys417-equivalent position in RaTG13.
The revised manuscript no longer includes discussion regarding the RaTG13-CoV spike.
12. How well does the WT SARS-CoV-1 spike construct (controlled for pre-fusion) bind ACE2 and behave in the pseudovirus entry experiment? Is it comparable to the SARS-CoV-2 K417V mutant? If it is, this would suggest that the two major switches for dictating the open/closed confirmations are predominantly due to the presence of the furin motif loop and the salt bridge. If not, this could point to other factors involved in contributing to the conformational differences between SARS-CoV-1 and -2.
In response to the first question, the functions of the SARS-CoV-1 and SARS-CoV-2 spikes were thoroughly compared in one of our earlier publications (PubMed ID 32376634). The present study is specifically concentrated on SARS-CoV-2 spike.
In the revised manuscript, we added the percentages of open and closed SARS-CoV-1 spike particles:
“SARS-CoV-1 spike predominantly assumes an open conformation (89% open and 11% closed) (28).”
We also noted that there may be other factors contributing to the conformations of coronavirus spikes:
“Besides residue 417, there are likely other molecular factors that influence the opening and closing of SARS-CoV-2 spike. Studies have shown that N-linked glycans on the spike and fatty acids bound to the spike both play a role in regulating its conformation (43, 44). As for protein-based factors, a D614G mutation that emerged later in the pandemic caused SARS-CoV-2 spike to favor the open conformation (45, 46). Furthermore, our earlier research indicated that three lysine residues kept SARS-CoV-2 Omicron spike in the open conformation (32).”
13. Lys417 has been identified as important for ACE2 binding. Can the authors comment on this, in the light of this new data.
The revised manuscript includes new data indicating that the K417V mutation decreases the direct binding of the RBD to human ACE2 (Figure 5). Thus, our study reveals that the K417V mutation has opposing effects on the spike’s function: it opens up the spike for better ACE2 binding while weakening the RBD's direct binding to ACE2.
14. Glycans and fatty acids have also been suggested to play a role in the open-to-closed transition. Can the authors comment on their potential roles in the light of this new data.
We have added the following discussion to the revised manuscript:
“Besides residue 417, there are likely other molecular factors that influence the opening and closing of SARS-CoV-2 spike. Studies have shown that N-linked glycans on the spike and fatty acids bound to the spike both play a role in regulating its conformation (43, 44).”
15. The illustration of the mechanism of the closed-to-open transition Figure S3A was difficult to interpret. Similarly, the interfaces between S1 and NTD were also difficult to glean from the figure. We would encourage the authors to make two separate figures (or at least, two panels in one figure) to illustrate this better. Also, a surface representation might work better to show the interface between S1 and NTD.
The revised manuscript no longer includes the packing analysis of the interfaces. Due to the relative mobility of the RBDs, an accurate packing analysis would require cryo-EM data of even higher resolutions. Therefore, the revised manuscript concentrates on insights gleaned from the overall cryo-EM structures, which are supported by a variety of biochemical and functional data. Collectively, our results reveal that residue 471 is the key regulator of SARS-CoV-2 spike's conformation.
16. Could the authors include more spike structures (i.e. closed form of D614G) for comparing the interfaces in Figure S3? Spike proteins features extensive conformational heterogeneity. Even within the same category of open or closed spike, further classification generates slightly different structures. It is not entirely clear what level of changes is significant.
The revised manuscript no longer includes the packing analysis of the interfaces. Due to the relative mobility of the RBDs, an accurate packing analysis would require cryo-EM data of even higher resolutions. Therefore, the revised manuscript concentrates on insights gleaned from the overall cryo-EM structures, which are supported by a variety of biochemical and functional data. Collectively, our results reveal that residue 471 is the key regulator of SARS-CoV-2 spike's conformation.
17. Line 99-101: The authors should quote A. C. Walls et al., Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell, (2020).
This citation has been added to the revised manuscript:
“We identified residue 417 as potentially a key difference between the two spikes: in the closed SARS-CoV-2 trimeric spike, Lys417 in the RBD from one spike subunit forms a hydrogen bond with the main chain of Asn370 in the RBD from another spike subunit, stabilizing the RBDs in the closed conformation (29) (Figure 1B).”
18. Line 136-137: Reference for the double proline/C-term foldon.
References have been added to the revised manuscript:
“To stabilize the recombinant spike ectodomain, we introduced six proline mutations into its S2 subunit to lock up its pre-fusion structure, introduced mutations to its furin motif to prevent it from being cleaved during molecular maturation, and added a C-terminal foldon tag to facilitate its trimerization (20, 31, 32).”
19. It would be helpful to include expected molecular masses in the legends of Figures 1C, 3B and 4B where expected bands should appear.
In the revised figure 2 legend, we added the following description:
“The expected molecular weights of SARS-CoV-2 spike monomer and S2 monomer are ~180 kDa and ~80 kDa, respectively.”
20. It would be very helpful to include a table of experimental outcomes that includes the protein conformations of all WT proteins (CoV-1, CoV-2, and RaTG13) and mutants, a summary of ACE2 binding and pseudovirus entry.
In the revised manuscript, we have omitted the supplementary table that cataloged the conformation of spikes from multiple coronaviruses, including RaTG13. We believe that such an extensive summary of the literature is more suited to a comprehensive review article. Instead, the revised manuscript narrows its comparison to SARS-CoV-2, SARS-CoV-1, and NL63-CoV, as all three of these viruses use ACE2 as their receptor but result in different patient symptoms. We have encapsulated the conformations of the other two spikes as follows:
“SARS-CoV-1 spike predominantly assumes the open conformation (89% open and 11% closed) (28).” “ NL63-CoV spike remains closed (100% closed) (26).”
We also note that:
“To date, several other studies also investigated the conformation of SARS-CoV-2 spike using cryo-EM (27, 29, 39-42), some of which gave different ratios of open and closed spikes probably due to differences in sample preparations and/or protein constructions. In this study, the two spike constructs only differ at residue 417 and the two spike samples were prepared using the same procedure. Importantly, our cryo-EM analysis is consistent with our extensive biochemical and functional approaches. These varied experimental methods complement one another, making this study one of the most thorough in examining the conformation of SARS-CoV-2 spike.”
21. In line 441, is "26,126 particles" correct? This number seems to refer to particles selected from 2D for the entire dataset.
Please see Figure 2—figure supplement1, Figure 2—figure supplement2, and Supplementary File 1 for the updated cryo-EM procedures.
Reviewer #1 (Recommendations for the authors):
1. To be able to comment on the molecular mechanism of close-to-open transition, the authors need to look at atomic models of the RBD captured in both closed and open states. The reviewers would like the authors to submit an open RBD structure. The data for this should already exist (4.6A K417V/furin motif deletion mutant 3D reconstruction presented in figure 2C). To get an idea of the general mechanism of the closed-to-open transition, the authors could build a (where appropriate, side chain-less model) of the 4.6A K417V/furin motif deletion mutant, for which the data already exists.
The revised manuscript centers on residue 417, which acts as a molecular switch that regulates the conformation of SARS-CoV-2 spike. It has omitted the data and discussion about the furin motif deletion mutant (Figure 1). Accordingly, the title of the manuscript has been changed to “Lys417 acts as a molecular switch regulating the conformation of SARS-CoV-2 spike protein”.
2. The authors should compare the open RBD structure with the FnM-deletion (100% closed) structure. In addition, they should make an internal comparison between the open and closed protomers in the "open" structure to gain an insight into the mechanism.
The revised manuscript centers on residue 417, which acts as a molecular switch that regulates the conformation of SARS-CoV-2 spike. It has omitted the data and discussion about the furin motif deletion mutant (Figure 1). Accordingly, the title of the manuscript has been changed to “Lys417 acts as a molecular switch regulating the conformation of SARS-CoV-2 spike protein”.
3. The way it is explained in the methods, only the highest resolution class is taken into consideration when the percentage occupancies are calculated for the open and closed states. This approach is inherently biased towards more structurally stable classes. Other open conformations might exist at lower resolutions. Ideally, the state occupancy percentages should be based on the total particle count.
For example, assuming that the open conformation is more unstable than the closed one, the real distribution could be 90% open and 10% closed, where only the 10% are selected and contribute to the high-resolution reconstruction. In this case, it'd be wrong to say that 100% of the particles are in the closed conformation when the data actually says that 100% of the high-resolution particles (and 10% of the total) are in the closed conformation. At the very least, the authors should discuss this aspect of data analysis and explain the implications for their hypothesis.
Please see Figure 2—figure supplement1, Figure 2—figure supplement2, and Supplementary File 1 for the updated cryo-EM procedures.
4. The authors should create a figure that explains their cryo-EM workflow for analysis in detail (representative micrograph, 2D classification, 3D classifications, etc). This figure should also contain local resolution plots and Euler angle distribution plots.
Please see Figure 2—figure supplement1 and Figure 2—figure supplement2 for the cryo-EM workflow.
5. The PDB validation and visual inspection of maps indicates that lots of residues/regions do not fit the map very well. Lots of side chains were built with no density supporting them. The authors should: a. remove segments of the model that are not supported by experimental density and b. re-build the backbone of the protein in parts of the protein where it does not fit the density well. This is particularly an issue because the authors discuss interaction networks in regions with poor density (Lys417 and its interaction partner are part of this region too).
The cryo-EM densities and the corresponding atomic models presented in this revision represent a substantial improvement over our previous submission. For detailed comparisons and data, refer to Figure 2—figure supplement1, Figure 2—figure supplement2, and Supplementary File 1.
6. The authors do not show a WT control in their mouse immunization experiments (figure 5). This control should be included.
The revised manuscript no longer includes mouse immunization data. Instead, it presents findings that the closed spike can evade the binding of many neutralizing antibodies and nanobodies (Figure 4).
7. The authors should discuss the Lys417 residue and its involvement in ACE2 binding.
The revised manuscript includes new data indicating that the K417V mutation decreases the direct binding of the RBD to human ACE2 (Figure 5). Thus, our study reveals that the K417V mutation has opposing effects on the spike’s function: it opens up the spike for better ACE2 binding while weakening the RBD's direct binding to ACE2.
Reviewer #2 (Recommendations for the authors):
Comments:
1) It would be important to know how well the WT SARS-CoV-1 spike construct (controlled for pre-fusion) binds ACE2 and behaves in the pseudovirus entry experiment. Is it comparable to the SARS-CoV-2 K417V mutant? If it is, this would suggest that the two major switches for dictating the open/closed confirmations are predominantly due to the presence of the furin motif loop and the salt bridge. If not, this could point to other factors involved in contributing to the conformational differences between SARS-CoV-1 and -2.
This revised manuscript focuses on residue 417 of SARS-CoV-2 spike. The comparison between the functions of SARS-CoV-1 and SARS-CoV-2 spikes was described in detail in one of our earlier publications (PubMed ID 32376634).
[Editors’ note: what follows is the authors’ response to the second round of review.]
Reviewer #1 (Recommendations for the authors):
The authors have submitted a streamlined and improved manuscript. I only have a few comments to add.
– My main concern is that the word "affinity" is not used properly in some parts of the paper. For example, in the abstract, the authors state "The net outcomes of this (K417V) mutation are to allow the spike to bind ACE2 more tightly…". This is not really what the data says- the mutation seems to increase the probability of "open" spike and therefore increases the probability of spike binding. In fact, the data shows that K417V binds less tightly to ACE2 than K417.
In the revised manuscript, we have changed “allow the spike to bind ACE2 more tightly” to “allow the spike to bind ACE2 with higher probability”.
In lines 116-118 the authors write "It has been shown that ACE2 only binds to the standing-up RBD in the open spike (28, 33), suggesting that open spike binds to ACE2 with higher affinity than the closed spike." A more accurate statement would be that the open spike conformation is necessary for ACE2 binding.
In the revised manuscript, we have changed “suggesting that open spike binds to ACE2 with higher affinity than does the closed spike” to “suggesting that the open spike conformation is necessary for ACE2 binding.”
This continues in lines 127-129 "The pull-down result showed that compared to K417-spike, more V417-spike molecules had been pulled down by ACE2, suggesting that V417-spike has a higher affinity for ACE2 (Figure 3A)." This experiment does not say anything about binding affinity, but it does suggest that the binding probability between the spike protein and ACE2 is increased in V417.
In the revised manuscript, we have changed “suggesting that V417-spike has a higher affinity for ACE2” to “suggesting that V417-spike had an increased probability of binding to ACE2”.
In the summary of findings in lines 184-187: "it weakens the RBD's ability to bind to ACE2 by eliminating a favorable interaction at the RBD/ACE2 interface, but it strengthens the trimeric spike's binding affinity for ACE2 by enabling more spike molecules to assume the open conformation." A more accurate statement would be that the V417 increases the probability of binding to ACE2 because it increases the probability of the open spike conformation.
In the revised manuscript, we have changed “it strengthens the trimeric spike's binding affinity for ACE2” to “it increases the trimeric spike's probability of binding to ACE2”.
https://doi.org/10.7554/eLife.74060.sa2Article and author information
Author details
Funding
National Institutes of Health (R01AI089728)
- Fang Li
National Institutes of Health (R01AI157975)
- Fang Li
National Institutes of Health (U19AI171954)
- Fang Li
National Institutes of Health (R01AI110700)
- Fang Li
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
This study was supported by funding from the University of Minnesota (to FL), NIH grants R01AI089728 (to FL), R01AI157975 (to FL), U19AI171954 (to FL, BL), and R01AI110700 (to FL).
Senior Editor
- Diane M Harper, University of Michigan, United States
Reviewing Editor
- Lejla Zubcevic, University of Kansas Medical Center, United States
Reviewers
- Lejla Zubcevic, University of Kansas Medical Center, United States
- Yi Ren, Vanderbilt University, United States
Version history
- Received: September 20, 2021
- Accepted: November 21, 2023
- Accepted Manuscript published: November 22, 2023 (version 1)
- Accepted Manuscript updated: November 24, 2023 (version 2)
- Version of Record published: December 4, 2023 (version 3)
Copyright
© 2023, Geng, Wan et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 348
- Page views
-
- 77
- Downloads
-
- 0
- Citations
Article citation count generated by polling the highest count across the following sources: Crossref, PubMed Central, Scopus.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Lys417 acts as a molecular switch that regulates the conformation of SARS-CoV-2 spike protein
eLife 12:e74060.
https://doi.org/10.7554/eLife.74060
Further reading
-
- Computational and Systems Biology
- Microbiology and Infectious Disease
Candida albicans, an opportunistic human pathogen, poses a significant threat to human health and is associated with significant socio-economic burden. Current antifungal treatments fail, at least in part, because C. albicans can initiate a strong drug tolerance response that allows some cells to grow at drug concentrations above their minimal inhibitory concentration. To better characterize this cytoprotective tolerance program at the molecular single-cell level, we used a nanoliter droplet-based transcriptomics platform to profile thousands of individual fungal cells and establish their subpopulation characteristics in the absence and presence of antifungal drugs. Profiles of untreated cells exhibit heterogeneous expression that correlates with cell cycle stage with distinct metabolic and stress responses. At 2 days post-fluconazole exposure (a time when tolerance is measurable), surviving cells bifurcate into two major subpopulations: one characterized by the upregulation of genes encoding ribosomal proteins, rRNA processing machinery, and mitochondrial cellular respiration capacity, termed the Ribo-dominant (Rd) state; and the other enriched for genes encoding stress responses and related processes, termed the Stress-dominant (Sd) state. This bifurcation persists at 3 and 6 days post-treatment. We provide evidence that the ribosome assembly stress response (RASTR) is activated in these subpopulations and may facilitate cell survival.
-
- Immunology and Inflammation
- Microbiology and Infectious Disease
Pyroptosis and apoptosis are two forms of regulated cell death that can defend against intracellular infection. When a cell fails to complete pyroptosis, backup pathways will initiate apoptosis. Here, we investigated the utility of apoptosis compared to pyroptosis in defense against an intracellular bacterial infection. We previously engineered Salmonella enterica serovar Typhimurium to persistently express flagellin, and thereby activate NLRC4 during systemic infection in mice. The resulting pyroptosis clears this flagellin-engineered strain. We now show that infection of caspase-1 or gasdermin D deficient macrophages by this flagellin-engineered S. Typhimurium induces apoptosis in vitro. Additionally, we engineered S. Typhimurium to translocate the pro-apoptotic BH3 domain of BID, which also triggers apoptosis in macrophages in vitro. During mouse infection, the apoptotic pathway successfully cleared these engineered S. Typhimurium from the intestinal niche but failed to clear the bacteria from the myeloid niche in the spleen or lymph nodes. In contrast, the pyroptotic pathway was beneficial in defense of both niches. To clear an infection, cells may have specific tasks that they must complete before they die; different modes of cell death could initiate these ‘bucket lists’ in either convergent or divergent ways.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}